

Initialement, les sarcomes d'Ewing ont été décrit comme des sarcomes à cellules rondes affectant les os.
Aujourd'hui, il est bien établi que les sarcomes d'Ewing partagent des traits communs avec les sarcomes extra-squelettiques comme les tumeurs primitives neuro-ectodermiques (PNET) et les tumeurs d'Askin. Toutes ces lésions ont une anomalie chromosomique en commun: la présence d'une translocation entre les longs bras des chromose 11 and 22 (t[11;22][q24;q12]). La tendance est donc de ne plus faire de distinction entre ces sarcomes à cellules rondes squelettiques et extra-squelletiques et de parler de tumeurs de la famille d'Ewing
Les sarcomes d'Ewing sont des sarcomes à cellules rondes affectant commence les os longs des enfants.
Ces tumeurs sont le plus souvent rencontrés chez les adolescents. Elles sont exceptionnelles après 25 ans [5-25ans].
Ces tumeurs peuvent survenir dans n'importe quel os.
Les tumeurs osseuses de Ewing sont le plus souvent recontrées dans la diaphyse (ou la région métadiaphysaire) des os longs (70% des cas). Les atteintes sarcomateuses confinées à la metaphyse sont moins fréquentes (moins de 10% des cas). Les tumeurs osseuses de Ewing centrés sur l'épiphyse sont rares (moins de 2% des cas).
Par odre de fréquence décroissante, les os longs atteints sont: le femur, l'ilium, le tibia, l'humérus, et la fibula. Les atteintes des os plats comme le sacrum ou la scapula, les côtes, les corps vertébraux sont moins fréquentes. Mais virtuellement, n'importe quel os peut être atteint comme le montre les différentes publications.
Ces tumeurs se manifestent le plus souvent localement par la présence d'une masse au voisinage et de douleurs osseuses.
Les tumeurs osseuses de Ewing sont des lésions très malignes et, au moment du diagnostic, dans 30% des cas, des métastases pulmonaires et osseuses sont déjà présentes.
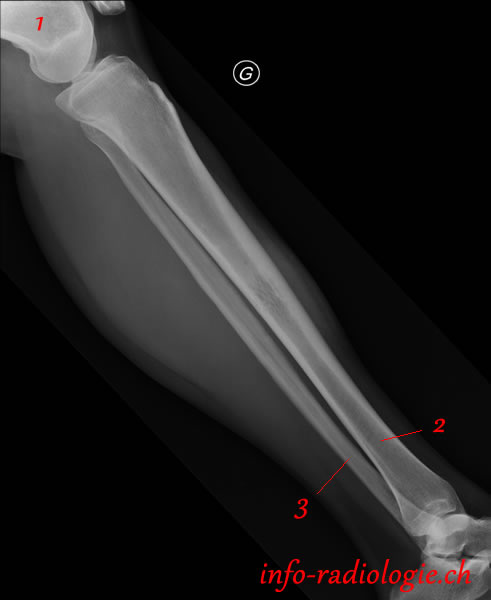
Le plus souvent, le sarcome d'ewing apparaît comme une lésion perméative (multiples petits trous), imtra médullaire, dans la diaphyse d'un os long chez un enfant. Plus rarement, la lésion peut être avoir un caractère mixte, c'est-à-dire lytique et sclérotique. Une apposition périostée plurilamellaires dite en bulbe d'oignon ou spiculée en rayons de soleil (lamelles perpendiculaires à la diaphyse) peut être présente.
L'IRM montre superbement l'envahissement tumoral endocanalaire et la masse circonférentiel des tissus mous. Cet examen est essentiel dans le bilan initial.
Ce jeune home de 39 ans, sans antécédent médical ou chirurgical, est envoyé par son médecin traitant pour investigations de douleurs osseuses et d'une masse des tissus mous autour du tibia. Selon le patient, la masse tibiale et la symptomatologie seraient apparues trois semaines avant le début des investigations.
Le bilan commence par de simple radiographies de la jambe. L'apparence des lésions impose une IRM. D'emblée, les investigations sont poursuivies par un scanner thoraco-adominal puis une biopsie chirurgicale de la lésion.
Remarques :
Sur un livre de référence (de langue anglaise), il est indiqué: «.... après 25 ans, la tumeur de Ewing ne doit plus être mis dans le diagnostic différentiel...... ». En médecine, on ne doit jamais dire jamais!
Sarcome de Ewing.
• Murphey MD1, Senchak LT, Mambalam PK, Logie CI, Klassen-Fischer MK, Kransdorf MJ. From the radiologic pathology archives: ewing sarcoma family of tumors: radiologic-pathologic correlation. Radiographics. 2013 May;33(3):803-31.
• Javery O, Krajewski K, O'Regan K, Kis B, Giardino A, Jagannathan J, Ramaiya NH. A to Z of extraskeletal Ewing sarcoma family of tumors in adults: imaging features of primary disease, metastatic patterns, and treatment responses. AJR Am J Roentgenol. 2011 Dec;197(6):W1015-22.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}